Hi! I was looking through the gentropy code, and I noticed, from the locus_breaker_clumping.py script, that there was p-value filtering done (line 68). Seeing as these results seem to feed directly into the susie-inf pipeline, I wanted to ask, are the summary statistics used as input into susie-inf filtered by p-value?
Thanks so much!
Hi! Thank you for reaching out.
Yes, we use a cut-off of 1e-8 to decide the SNPs that construct the Locus object, which is then fed into the susie fine-mapper.
I hope this helps!
Best,
Annalisa
Thanks so much! So just to confirm that I’m understanding correctly, the variants on which fine-mapping (via Susie-Ind) is conducted are filtered by this pvalue?
Appreciate all the help!
Dear @abhukku
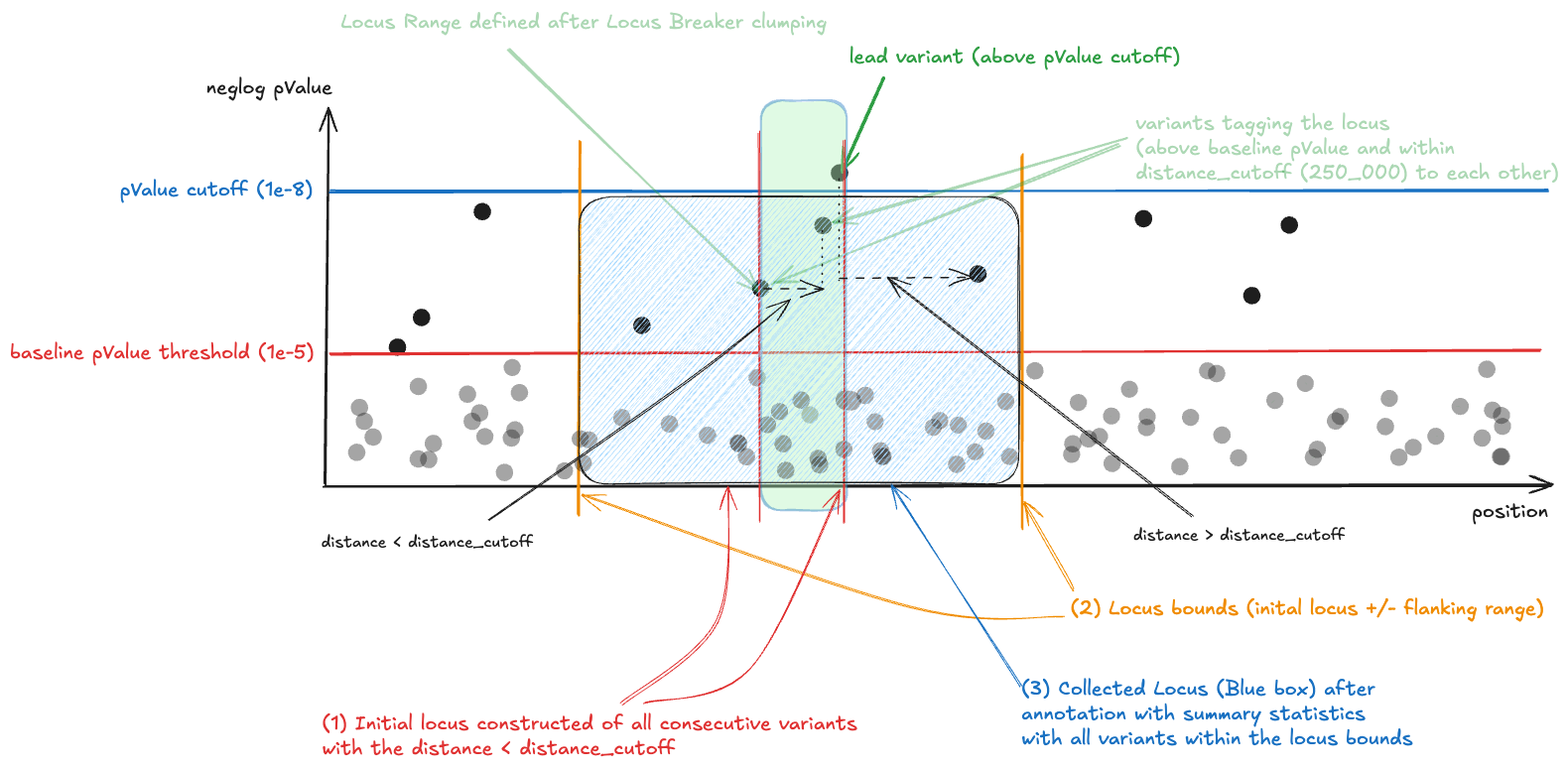
The process is slightly more complex then just filtering summary statistics variants by the p-value. Full explanation is available at the Locus Breaker documentation.
When we break the summary statistics into loci we define two p-value thresholds:
-
baseline_pvalue_cutoff (1e-5) - it is used for initial filtering of the summary statistics to obtain all valuable variants that can generate loci.
-
pvalue_threshold (1e-8) - any variant below this threshold can be seen as a locus lead variant. If a locus does not have any variant with p-value below this threshold, it is dropped.
When constructing the locus object we do following procedure (simplified version)
First we filter out all variants from summary statistics when p-value > baseline_pvalue_cutoff, then we:
- Iterate over the variants and compare each consecutive variant distances, based on that we collect all sets of variants with when the distance between two consecutive variants is below distance_cutoff (250.000 bp). This is the initial locus (green box at the schema).
- Assign the locusStart and locusEnd (locus bounds) by subtracting/adding the flanking range (100.000bp) to the initial locus bounds (blue box at the schema)
- Rank the locus by p-value and filter out loci that do not have at least 1 significant variant (filter out loci without variant with p-value below pvalue_threshold)
After we get the locus object (lead variant, locusStart and locusEnd) we annotate each locus with corresponding summary statistics based on the locus bounds. All variants are preserved in the locus object. The variants are only limited to locus bounds, no p-value filter is applied on the locus object.
This is the schematic representation of what is happening within the locus breaker.
I encourage you to have a look at the LocusBreakerClumpingStep to see what are the other computations that we use to transform the summary statistics to Locus object
I also encourage you to take a look into the code defined in orchestration to see the exact parameters used to define June release.
2 Likes